

| 导读 |
伴随诊断在1998年首次被FDA批准,2021年它将占据体外诊断市场14%的份额,能够提供有关患者针对特定治疗药物/生物制品的治疗反应的信息。作为药企和诊断试剂开发商,如何更好地进行“伴随诊断”的合作开发?不妨看看FDA最新发布的长达48页的“葵花宝典”。
|

(一)什么是伴随诊断?
伴随诊断(Companion Diagnostics,简称CDx)之路始于1998年FDA批准的抗癌药物赫赛汀(Herceptin),它包括对某种药物进行的同步诊断和包括对某种生物制品进行同步诊断两种类型。
按照目前赫赛汀每次治疗均价2.5万左右计算,而每次接受肿瘤治疗药物测试价格300元/次(福建省卫计委公布最新价格)。因此,比起盲目服用高昂的药物以及忍受冗长的治疗周期,伴随诊断技术有助于快速判断抗癌药物方案是否适合具体的患者。
根据美国FDA官方网站的定义:伴随诊断疗法是一种医疗器械,作为一种体外诊断(IVD)技术,能够提供有关患者针对特定治疗药物/生物制品的治疗反应的信息。这种测试有助于判断某种治疗产品的受益患者群体、严重副作用群体和潜在风险群体。
具体而言,伴随诊断可以做以下三件事情:
1)确定哪些患者是某种特定疗法的最可能受益群体;
2)确定患者在接受某种特定疗法后可能产生的严重的副作用;
3)监控与特定治疗对治疗的反应产品调整治疗的目的来实现改进的安全性或有效性。
值得一提的是,如果诊断测试是不准确的,那么基于测试结果的治疗决策可能不是最优的。
(二)美国FDA发布最新的药物/诊断共同开发指导原则(草案)
早在2014年7月31日,FDA发布了《体外伴随诊断测试产业指南》,旨在帮助制药企业在早期的药物研发中确定是否需要伴随诊断以及是否需要制定药物和伴随诊断的联合开发计划。

2016年7月15日,FDA公布了指南草案——《体外伴随诊断设备与治疗产品的共同开发指导原则》,该文件旨在成为一份实用指南,以帮助药企和诊断试剂商更好地进行“药物-诊断”合作开发。
FDA表示,这份指南草案将在其官网上公示90天至10月13日,公民可以通过www.regulations.gov.网站进行反馈留言;也可以通过写信给FDA的文档管理部进行反馈留言。
这份文档究竟说了些啥呢?生物探索编辑通过通读文档,带你提前了解。
(三)《指导原则》的大纲内容
I.序言 .............................................................................................................4
II.背景 ............................................................................................................6
III. 共同开发过程的原则 ................................................................................7
A. 一般原则 ....................................................................................................8
B. 体外诊断和治疗产品的监管条例 .............................................................9
1. 风险评估和医疗器械临床试验申报要求 ....................................................10
2. 在研药物或生物制品相关的IVD信息递交 ..................................................13
3. 共同开发试验中IVD产品的医疗器械临床试验申报要求 ..........................14
C. 潜在合作发展项目的IVD验证计划 .........................................................15
1. 研究性IVD试剂在治疗产品试验前的分析验证预期 ...................................15
2. IVD试剂的新型用途 ........................................................................................16
3. 治疗产品早期临床试验阶段的IVD试验 ........................................................16
4. 研究试剂仅作测试系统的一部分 ...................................................................17
5. 筛选可用于治疗产品临床试验的IVD .............................................................18
6. 分析前程序和测试协议 ....................................................................................19
7. 分析验证研究的前瞻性规划 ............................................................................19
D. 治疗产品临床试验设计注意事项 ...............................................................20
1. 早期治疗产品开发的一般注意事项 .................................................................21
2. 晚期治疗产品开发的一般注意事项 .................................................................22
3. 预后及预测标记物 .............................................................................................24
4. 前瞻性回顾的方法 .............................................................................................25
5. 确定预期人群的注意事项 .................................................................................26
E. 晚期治疗产品的IVD开发注意事项 ...........................................................28
1. 训练样本集与验证样本集 .................................................................................29
2. 实验设计的变量影响 .........................................................................................29
3. IVD 衔接性试验 .................................................................................................30
4. 特殊方案的评估 .................................................................................................31
F. 同期营销授权计划 .....................................................................................32
1. 协调审核时间表 .................................................................................................32
2. 什么条件下不能进行同期营销授权 .................................................................37
3. 一种IVD伴随诊断在获得营销授权前的装运与验证 ......................................37
G.标签注意事项 ............................................................................................38
1. 试验性IVD伴随诊断的要求 ..............................................................................38
H.上市前注意事项 .........................................................................................40
附录1:共同开发过程的关键点 ......................................................................41
附录2:主要样本处理的注意事项 ...................................................................43
附录3:递交PMA前的BIMO信息 ..................................................................46
录4:授权书 .................................................................................................47

(四)《指导原则》的核心内容
在这份长达48页的《体外伴随诊断设备与治疗产品的共同开发指导原则》(草案)中,最重要的部分莫过于第三大部分——共同开发过程的原则,其中包括体外诊断和治疗产品的监管条例 、潜在合作发展项目的IVD验证计划、治疗产品临床试验设计注意事项、晚期期治疗产品的IVD开发注意事项、标签注意事项以及上市前注意事项等。
其中,关于治疗产品临床试验与IVD的产品验证试验中,需要注意的事项尤为重要,本文仅用以下两个图表进行解读。在进行伴随诊断产品开发及申报过程中,可以参照大纲内容进行文档索引。
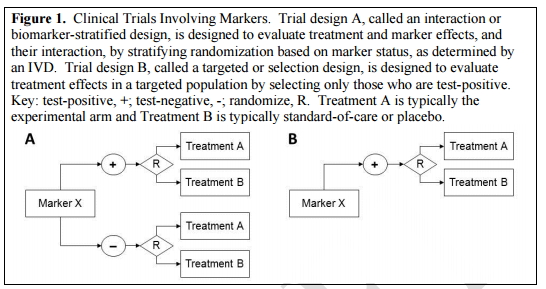
注:图中“+”指阳性,“-”指阴性,R指随机,A代表实验组,B代表安慰剂组
众所周知,在开发伴随诊断产品时,离不开对标记物进行临床试验的分析与验证。
图1A叫做“生物标志物的分层设计”或“生物标志物的相互作用”,用以评价治疗产品与标记物的效果以及它们之间的相互作用,该方法作为一种IVD产品测试,是基于标志物随机状态下的分层试验;
图1B叫做“靶向设计”或“选择设计”,旨在评估特定人群中,谁能够对治疗方法产生阳性效果的测试。
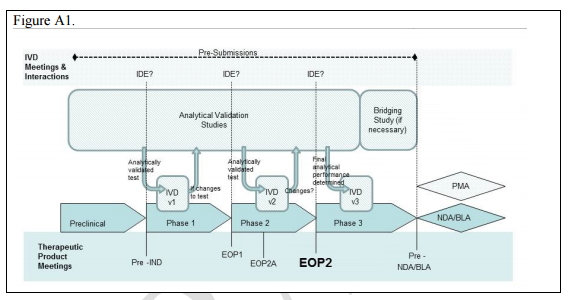
共同开发过程的关键点
毋庸置疑,一种治疗方法的伴随诊断的高效合作方式必须是建立在:两个产品研发项目团队的积极协作,甚至包括FDA系统所有相关部门间的合作与相互交流。
如图1所示:在某个药物/生物制品从临床前研究到NDA/BLA(新药上市许可/生物制品许可申请)过程中,先后需要经过临床前研究,1、2、3期临床试验。
在这个过程中,相应的体外诊断测试也需要同步进行(见图中的IVD v1、IVD v2、IVD v3)。其中针对在临床前研究的完成时(Pre-IND)、1期临床试验结束点(EOP1)和2期临床试验结束点(EOP2)所开发的伴随诊断测试都需要进行医疗器械临床试验申报(IDE)。
在上述的过程中,如何设计相应控制点的目标?如何衡量出现的偏差?谁应对哪些失误负责?哪些信息反馈价值最大、最经济实用等等。具体说来,企业在设计试验时就需要要制定一些客观的标准。
(五)FDA已经批准的28款伴随诊断产品
根据生物探索的前期统计(截止到2016年5月24日),FDA已经批准了一些伴随诊断检测。这些检测由罗氏、雅培、QIAGEN、DAKO等公司推出,主要利用qPCR、原位杂交、免疫组化等方法筛查一些肿瘤相关突变,以协助医生选择适当的疗法。

备注:NDA指新药申请;BLA指生物制剂许可申请;PMA指上市前申请许可;IU指预期用途;IFU指使用适应症。
(六)展望:伴随诊断,体外诊断市场发展最快的领域
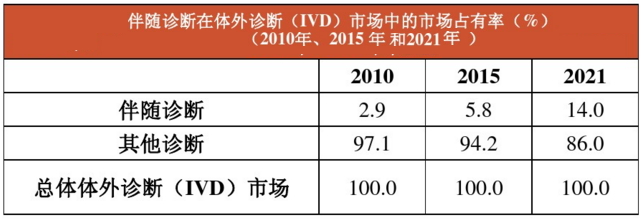
由上图可见,伴随诊断是一个新兴市场,预计到2021年,伴随诊断将占整个体外诊断(IVD)市场14%的份额。
目前已经有越来越多的医疗器械(IVD)公司开始与制药企业展开合作,为后者的在研药物/生物制品开发伴随诊断试剂盒。
作为精准医疗的重要分支,伴随诊断是对于预测患者针对特定药物的治疗反应至关重要。
对尚未或正在准备开展伴随诊断合作,拓宽新兴市场的药企&诊断公司而言,仔细研究FDA发布的这份长达48页的“葵花宝典”,必将让“研发——试验——上市”过程更加顺利。
“葵花宝典”下载地址:
Principles for Codevelopment of an In Vitro Companion Diagnostic Device with a Therapeutic Product
